

Enfermedades Raras (ER): Síndrome de Holt-Oram (HOS)
- 21 feb 2018
- 2 Min. de lectura
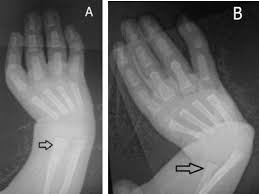
El síndrome de Holt-Oram es una enfermedad de origen genético que se considera una variante del síndrome corazón mano. Produce malformaciones que afectan a varios órganos, principalmente extremidades superiores y corazón. Es muy poco frecuente por lo que se incluye dentro de las Enfermedades Raras.
Los síntomas principales consisten en malformaciones de los huesos de la mano y extremidades superiores, incluyendo focomelia, pulgares ausentes o con 3 falanges en lugar de las 2 habituales y falta de desarrollo del radio que ocasiona longitud desigual de los brazos.
En el corazón puede existir comunicación interauricular, comunicación interventricular y en ocasiones arritmias como fibrilación auricular.
Otro tipo de anomalías que se han descrito incluyen anomalías craneofaciales, axilares, traqueales, vertebrales y de las extremidades inferiores, así como sordera, situs inversus abdominal y anomalías renales, pero en muchos casos estos hallazgos reflejan fenocopias del síndrome en vez del propio HOS.
Está provocado por una mutación en el gen TBX5 situado en el cromosoma 12 humano (12q24.1). Suele ser debido a una mutación nueva en dicho gen, en este caso no existen antecedentes familiares. Cuando uno de los padres está afectado, existe un 50% de probabilidades de que un hijo determinado presente la enfermedad, por ser las transmisión autosómica dominante. El gen TBX5 codifica un factor de transcripción que regula la expresión de otros genes implicados en el desarrollo del corazón y los miembros superiores.
Las pruebas prenatales se basan en el análisis de ADN procedente del feto obtenido tras amniocentesis y biopsia corial, y pueden ser útiles para confirmar los hallazgos de la ecografía y ecocardiografía en familias con una mutación HOS conocida.
El manejo es multidisciplinar e involucra a genetistas, cardiólogos, cirujanos ortopédicos y ortopédicos pediátricos, así como redes de apoyo social. Los pacientes con bloqueo cardíaco avanzado pueden requerir un marcapasos permanente. Se recomienda un ecocardiograma cada cinco años si existen defectos cardíacos. Se recomienda realizar electrocardiogramas (ECG) anuales a los adultos.
El pronóstico es variable. El manejo funcional en la vida cotidiana del paciente estará limitado por el tipo y la gravedad de las anomalías de las extremidades superiores que presente. La esperanza de vida depende de la gravedad de las anomalías cardíacas.
Os invito a seguir la colmena de Enfermedades Raras, donde podréis compartir todo aquello relacionado con estas enfermedades e informaros de las numerosas ER.
Fuentes: Wikipedia, FEDER, Orphanet
Fernando Santa Isabel
______________________________
Mi Twitter
Mi LinkedIn
Mi beBee
Mi página web
El contenido del presente artículo es de carácter general y tiene una finalidad meramente informativa, sin que se garantice plenamente el acceso a todos los contenidos, ni su exhaustividad, corrección, vigencia o actualidad, ni su idoneidad o utilidad para un objetivo específico. Toda la información que se ofrece a través de este artículo no sustituye, en ningún caso, un asesoramiento sanitario cualificado. Así mismo declino toda responsabilidad sobre las consecuencias que un mal uso de este contenido pueda tener en la salud de los pacientes.


























Comentarios